
WISP1 and TLR4 Signaling in Ventilator-induced Lung Injury (VILI)
PI: Li-Ming Zhang, MD
Collaborators: Timothy R. Billiar, MD (George Vance Foster Professor and Chair, Department of Surgery) and Bruce R. Pitt, PhD (Immediate Past Chair, Department of Environmental and Occupational Health)
The Zhang group’s previous unbiased genome-wide association studies in a genetically diverse panel of 23 mouse strains identified WNT1-inducible signaling pathway protein 1 (WISP1) as playing an important role in a murine VILI model. In addition, they identified that innate immune signaling via Toll-like receptor 4 (TLR4) plays a critical role in the pathogenesis of VILI and that stretch-induced WISP1 expression and its pro-inflammatory effect were TLR4-dependent.
Sepsis is a common predisposing factor for acute respiratory distress syndrome, and many patients with this condition require mechanical ventilation. Although both sepsis and mechanical ventilation signal via TLR4 as Dr. Zhang’s research group and others have previously shown, the molecular determinants underlying the activation of TLR4 signaling in the latter condition with different tidal volumes are unknown.
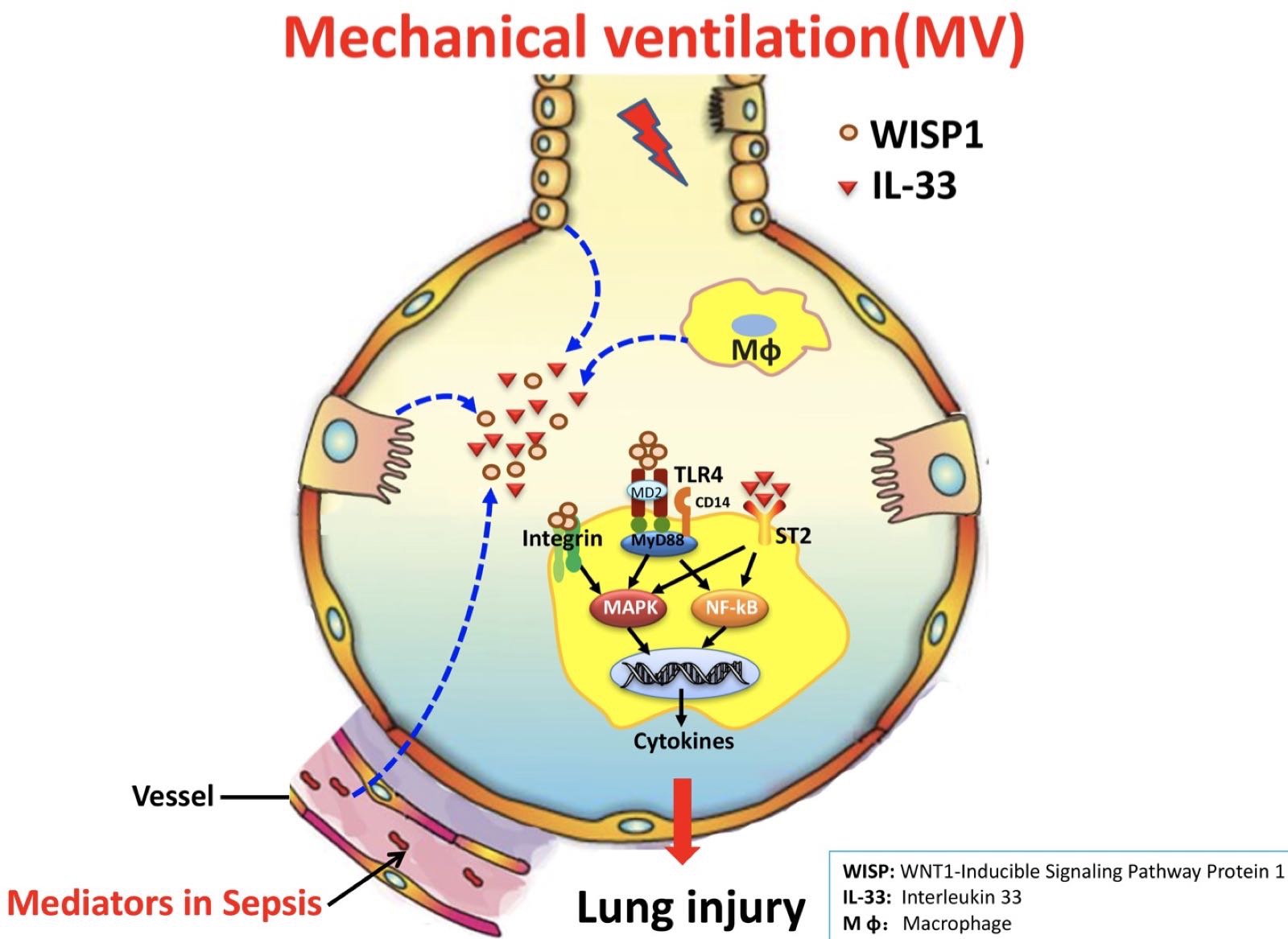 Dr. Zhang’s research group proposes to further investigate the role of WISP-1, αvβ3 and β5 integrin, IL-33, and TLR4 signaling in their newly-developed and highly relevant preclinical model of (polymicrobial) sepsis and mechanical ventilation (two-hit model: MV+CLP-sepsis) in intact mice. They and their colleagues noted that the matricellular protein WISP1 contributes to TLR4-mediated sterile and infectious acute lung injury (ALI). The role of integrins in mediating the WISP1 effect was apparent in the arginylglycylaspartic acid (RGD)-sensitive nature of cecal ligation and puncture (CLP)-induced ALI and a component that could be ascribed to WISP1-integrin β6 interaction via neutralizing antibodies. Although sepsis is the primary cause of respiratory failure, requiring the supportive measures of mechanical ventilation, and mechanical ventilation itself may cause iatrogenic ALI, few reports have combined these stimuli in a calibrated double hit ALI model. Accordingly, they explored the role of WISP1 and integrin β5 and IL-33 in the underlying mechanisms of ALI during mechanical ventilation in a murine model (CLP) of polymicrobial sepsis.
Dr. Zhang’s research group proposes to further investigate the role of WISP-1, αvβ3 and β5 integrin, IL-33, and TLR4 signaling in their newly-developed and highly relevant preclinical model of (polymicrobial) sepsis and mechanical ventilation (two-hit model: MV+CLP-sepsis) in intact mice. They and their colleagues noted that the matricellular protein WISP1 contributes to TLR4-mediated sterile and infectious acute lung injury (ALI). The role of integrins in mediating the WISP1 effect was apparent in the arginylglycylaspartic acid (RGD)-sensitive nature of cecal ligation and puncture (CLP)-induced ALI and a component that could be ascribed to WISP1-integrin β6 interaction via neutralizing antibodies. Although sepsis is the primary cause of respiratory failure, requiring the supportive measures of mechanical ventilation, and mechanical ventilation itself may cause iatrogenic ALI, few reports have combined these stimuli in a calibrated double hit ALI model. Accordingly, they explored the role of WISP1 and integrin β5 and IL-33 in the underlying mechanisms of ALI during mechanical ventilation in a murine model (CLP) of polymicrobial sepsis.
Their results suggest that WISP1-αvβ3 integrin signaling is a critical immune modulator in TLR4 pathways in macrophages and may be an important contributor to TLR4/CD14-mediated inflammation in polymicrobial sepsis-induced lung injury. Their results also showed that mechanical ventilation with normal tidal volume increased both WISP1 production and integrin β5 expression two-fold in intact lung after CLP and signaled through TLR4-MyD88 to aggravate sepsis-induced lung injury, while mechanical ventilation with low tidal volume produced a protective effect on lung injury induced by CLP/sepsis.
Their recent results showed that the IL-33-ST2 pathway plays a dominant role in lung injury observed when moderate tidal volume (MTV, 10ml/kg) is used within the first six hours following the onset of sepsis. The strong induction of IL-33 by MTV and conversely, the suppression of IL-33 by low tidal volume (LTV 6ml/kg), further confirm that IL-33 expression and signaling is very much dependent on the degree of mechanical stretch. Their results support the use of protective ventilation strategies when mechanical ventilation is required for the management of intra-abdominal sepsis.
In addition, their recent study explored the potential role of non-canonical Wnt signaling in the pathogenesis of VILI and found that six hours of mechanical ventilation induced lung injury in a volume-dependent fashion (LTV: 6 ml/kg or MTV: 12 ml/kg) in a previously reported sensitive strain (A/J) of mice. MTV ventilation increased the protein levels of WISP1, phospho glycogensynthase kinase, β-catenin, phosphorylated Ras homolog gene family, member A, and phosphorylated C-Jun N-terminal kinase (p-JNK). Inhibition of rho-associated, coiled-coil-containing protein kinase 1 by Y27632 and JNK by SP600125 attenuated MTV-induced lung injury and decreased the protein levels of components in the non-canonical Wnt signaling pathways, including WISP1.
Significant Results
They confirmed that TLR4 and CD14 are critical in transducing CLP-mediated ALI (including the elevation of intrapulmonary WISP1) and demonstrated that intrapulmonary αvβ3 is increased by polymicrobial sepsis in a TLR4, CD14-dependent fashion. Comparison of cultured macrophages revealed that: a) WISP1 by itself increased the release of tumor necrosis factor alpha (TNF-α) from RAW264.7 cells (with baseline expression of αvβ3) in an RGD-sensitive fashion; but b) primary cultures of peritoneal macrophages (PMø) required activation of TLRs to induce de novo synthesis of αvβ3, enabling WISP1 to stimulate release of TNF. The specific requirement for β3 integrin was apparent when the effect of WISP1 was lost in PMø isolated from β3-/- mice. WISP1 enhanced TLR4-mediated extracellular-signal-regulated kinase (ERK) signaling; U0126 (an ERK inhibitor) blocked lipopolysaccharide (LPS)-induced β3 integrin expression; and WISP1 enhanced TNF-α release.
They also showed that LPS induced upregulation of integrin β5 through the TLR4-MyD88 signaling pathway. They further demonstrated that mechanical ventilation with normal tidal volume significantly aggravated mild sepsis-induced lung injury and inflammation responses, whereas in TLR4-null mice, intratracheal administration of WISP1 antibody or integrin β5 antibody significantly attenuated lung injury. These findings were recapitulated in mouse PMø in vitro after LPS and WISP1 recombinant protein in a sequential stimulation. The enhanced integrin β5 expression in murine PMø augmented the production of cytokines and chemokines, which was accompanied in the amplified polymorphonuclear leukocyte migration into the lung. However, these enhanced inflammatory responses vanished in PMø obtained from TLR4-null mice, and furthermore, inhibition of integrin β5 expression by applying the integrin β5 siRNA decreased the inflammatory response.
In addition, they showed that mild lung injury secondary to sepsis (CLP; 12 h) can sensitize intact mouse lung to subsequent moderate tidal volume (10 ml/kg) ventilation injury (manifested as increased alveolar capillary permeability, lung injury, neutrophil sequestration, and synthesis and release of cytokines and chemokines) for prolonged periods of ventilation (6 h). This two-hit model is TLR4-sensitive and shows important pro-injury and inflammatory contributions from both WISP1 and integrin β5. In isolated PMø, we further defined the nature of WISP1 signaling and identified a requisite TLR4-dependent activation of αVβ5 and MyD88-NF-kB pathway inflammatory mediator biosynthesis.
Their study further confirms that mechanical ventilation induces lung injury paralleled with increasing WISP1 expression. Non-canonical Wnt signaling involved in mechanical VILI and the suppression of the non-canonical Wnt signaling pathway significantly alleviated VILI. WISP1 expression is regulated by the non-canonical Wnt signaling pathway in A/J mice. The modulation of both WISP1 and Wnt signaling may provide novel therapeutic strategies for VILI.
Key Outcomes
- CLP, WISP1-αvβ3 integrin, and TLR4 signaling: Collectively these data suggest that WISP1-αvβ3 integrin signaling is a critical immune modulator in TLR4 pathways in macrophages and may be an important contributor to TLR4/CD14-mediated inflammation in polymicrobial sepsis-induced lung injury.
- CLP, mechanical ventilation, WISP1-αvβ5 integrin, IL-33, and TLR4: These results indicate that mechanical ventilation with MTV (10ml/kg) increased both WISP1 and IL-33 production and integrin β5 expression two-fold in intact lung after CLP and signaled through TLR4-MyD88 to aggravate sepsis-induced lung injury, while mechanical ventilation with LTV (6 ml/kg) suppressed WISP1 and IL-33 production to attenuate sepsis-induced lung injury.
- Non-canonical Wnt signaling participates in VILI by modulating WISP1 expression that we previously noted was critical for development of VILI. The non-canonical Wnt signaling pathway could provide a preventive and therapeutic target for VILI.
Publications
- Chang J, Xia Y, Wasserloos K, Deng M, Blose KJ, Vorp DA, Turnquist HR, Billiar TR, Pitt BA, Zhang MZ, Zhang LM. Cyclic stretch induced IL-33 production through HMGB1/TLR-4 signaling pathway in murine respiratory epithelial cells. PLoS One. 2017 Sep 12;12(9):e0184770. doi: 10.1371/journal.pone.0184770. eCollection 2017.
- Ding X, Tong Y, Jin S, Chen ZX, Li TL, Billiar TR, Pitt BR, Li Q & Zhang LM. Mechanical ventilation enhances sepsis-induced lung injury: role of WISP1-αvβ5 integrin pathway in TLR4 mediated inflammation and injury. Critical Care 2018 DOI.10.1186/s13054-0182237-0
- Ding X, Jin S, Shao Z, Xu L, Yu Z, Tong Y, Chen Z, Turnquist H, Pitt BR, Billiar TR, Zhang LM, Li Q. The IL-33-ST2 Pathway Contributes to Ventilator-Induced Lung Injury in Septic Mice in a Tidal Volume-Dependent Manner. Shock. 2018 Sep 5. DOI: 10.1097/SHK.0000000000001260. [Epub ahead of print]
- Xia XF, Chang J, Yang JF, Ouyang W, Pitt BR, Billiar TR, Zhang LM. Non-canonical Wnt signaling contributes to ventilator-induced lung injury through upregulation of WISP1 expression. Int J Mol Med, 2018, in press.